Video Article Open Access
How Protonation and Deprotonation Affect Weakening of Epoxy Resin and Silane Coupling: First-Principles Simulation Based on Free Energy Calculation
Shuji Ogata*, Masayuki Uranagase
Graduate School of Engineering, Nagoya Institute of Technology, Nagoya, 466-8555, Japan
Vid. Proc. Adv. Mater., Volume 3, Article ID 2208316 (2022)
DOI: 10.5185/vpoam.2022.08316
Publication Date (Web): 23 Dec 2022
Copyright © IAAM
Graphical Abstract
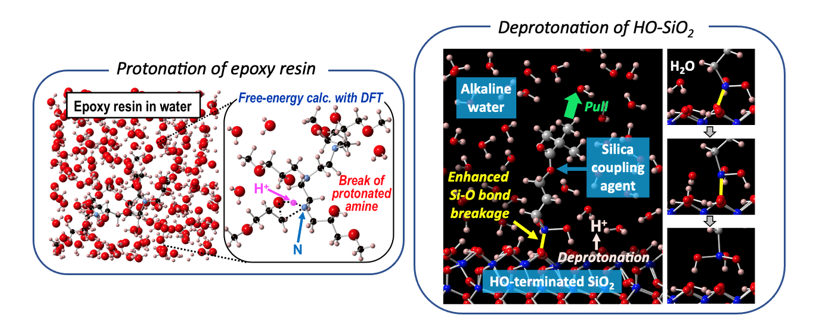
Abstract
Protonation and deprotonation of adhesion interface under wet conditions often result in degradation. We unveil their microscopic mechanisms from the first-principles simulation.
We investigated the protonation of the amine group in epoxy resins prepared using amine-based curing agents by theoretical methods [1]. The density functional theory (DFT)-based free-energy calculations of the corresponding deprotonation sub-reactions showed that the amine group of the epoxy resin is protonated at equilibrium depending on the location of the amine group when the epoxy resin is embedded in water under standard conditions. Additional DFT calculations demonstrate that the energetic barrier for breaking the ether bond of the epoxy resin is lowered by about 0.6 eV as a result of the cooperative effect of H2O dissociation and that the transition state energy for breaking the amine group bond is lowered by about 0.4 eV after the protonation of the amine group. Comparing the transition state energies, we predict that the bond breakage of the protonated amine groups is the principal process causing the weakening of epoxy resins under wet conditions.
Silane coupling is commonly used to connect organic polymers to inorganic substrates for surface modification and composite material fabrication. It is known that the covalent bonds that form between the silane coupling agent (SCA) and hydroxylated SiO2 weaken under alkaline conditions. In the present work, we theoretically investigated the microscopic mechanisms of this phenomenon by combining first-principles calculations of the deprotonation free energy with DFT, bond-breaking transition-state barrier energy calculations, and DFT-based molecular dynamics (MD) simulations [2]. We found that the following occurs in alkaline conditions: (i) the terminal –OH of SiO2(001) is significantly deprotonated, while the –OH group of the SCA is hardly deprotonated; (ii) the transition-state barrier energy to break the Si–O bond between the SCA and SiO2(001) becomes less than 1 eV with the assistance of H2O dissociation when 50% of the terminal –OH groups on SiO2(001) are deprotonated; and (iii) the predicted Si–O bond-breaking reactions occurred in DFT-MD simulations with realistic settings.
Keywords
Protonation; deprotonation; DFT; epoxy resin; silane coupling.
References
1. S. Ogata, M. Uranagase, Y. Takahashi, T. Kishi, J. Phys. Chem. B, 2021, 125, 8988.
2. S. Ogata, M. Uranagase, J. Phys. Chem. C, 2021, 125, 2209.
Video Proceedings of Advanced Materials

Upcoming Congress
