Video Article Open Access
Computational discovery of 2D materials: a fundament study of boron sheets and phosphide binary compounds
Ming Yu*, Congyan Zhang, Cheno B Kah
Department of Physics and Astronomy, University of Louisville, Louisville, Kentucky, USA
Vid. Proc. Adv. Mater., Volume 1, Article ID 2020-0810 (2020)
DOI: 10.5185/vpoam.2020.0810
Publication Date (Web): 06 Aug 2020
Copyright © IAAM
Graphical Abstract
Abstract
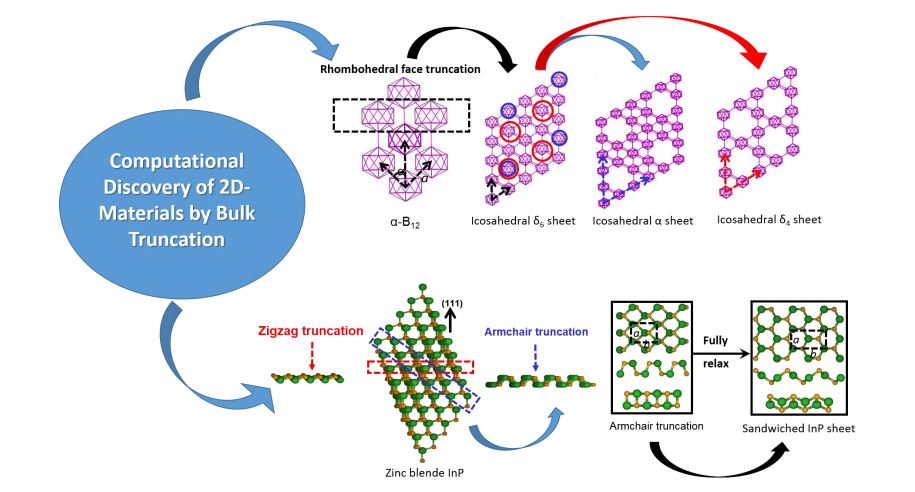
Accelerating the discovery of low-dimensional materials having improved properties and advanced capabilities are essential in the 21st century to enable future development of nanotechnology in challenge applications such as flexible electronics, portable sensors, solar panels, photodiodes, phototransistors, tunneling devices, etc. Experimental and computational simulating methodologies, as well as machine learning developed recently, are mainstream routes to explore unknown nanomaterials. In this lecture, I will focus on the computational discovery of low-dimensional materials employing first-principles and semi-empirical methods. There are serval routes (e.g., data mining route, global optimization route, chemical substitution route [1]) in predicting new nanomaterials. We are interested searching novel two-dimensional (2D) structures, which have no 3D vdW counterparts. Predicting such 2D layered structures requires physical intuitions. Most of them are predicted by chemical substitution route from the graphene-like honeycomb framework [2]. Alternatively, we explored unknown 2D layers by bulk truncations along certain orientations in order to keep their preferential chemical bonding nature exhibited in their crystalline structures. In this way, we not only found the same 2D structures predicted from the honeycomb framework, but also discovered new 2D allotropes. Two examples will be discussed: (1) low dimensional boron structures based on icosahedron B12 [3] and (2) sandwiched 2D phosphide binary compounds sheets [4]. The 2D icosahedral boron sheets (referred as icosahedral α, δ6, and δ4 sheets) that contain icosahedra B12 as their building units were obtained by rhombohedral face truncation from bulk α-B12. These novel 2D icosahedral structures exhibit interesting bonding and electronic properties. Specifically, the three-center, two-electron bonding between icosahedra B12 of the boron bulk transforms into a two-center bonding in these new allotropes of boron sheets. In contrast to the previously reported buckled α and triangular boron monolayer sheets [5], these new allotropes of boron sheets form a planar network. Their electronic properties show semiconducting nature for the icosahedral δ6 and δ4 sheets, and a nearly gapless feature for the icosahedral α sheet. On the other hand, atomic layers of GaP and InP binary compounds with unique anisotropic structural, electronic, and mechanical properties have been predicted by the armchair truncation from the zinc blende GaP and InP along (111) orientation. After fully relaxation, these new members of phosphide binary compounds family stabilize to a sandwiched 2D crystalline structure with orthorhombic lattices symmetry and high buckling of 2.14 Å-2.46 Å. Their vibration modes are similar to those of phosphorene with six Raman active modes ranging from ~80 cm-1 to 400 cm-1. The speeds of sound in their phonon dispersions reflect anisotropy in their elastic constants, which was further confirmed from their strong directional dependence of Young’s moduli and effective nonlinear elastic moduli. They show wide bandgap semiconductor behaviour with fundamental bandgaps of 2.89 eV for GaP and 2.59 eV for InP, respectively, even wider than their bulk counterparts. Such bandgaps were found to be tunable under the strains. In particular, a direct-indirect bandgap transition was found under certain strains along zigzag or biaxial orientations, reflecting their promising applications for the strain-induced bandgap engineering in nanoelectronics and photovoltaics.
Keywords
Computational discovery, Bulk truncation, Boron icosahedral structures, Sandwiched phosphide binary compounds sheets.
Acknowledgement
The authors are grateful for the computing resource support from the Cardinal Research Cluster at the University of Louisville.
References
Biography
My research area is condensed matter physics and computational nanomaterials science. My current research interests cover (1) Methodology Development of a semi-empirical Hamiltonian for studying large scale and complex systems; (2) First-principles calculations to understand, predict, and characterize structural, electronic, chemical, and mechanical properties of novel nanostructures; and (3) Applications for the next generation of energy storage, nanosensors, nanomaterial engineering, intercalations, and functionalized nanomaterials. Some of my impactful recent research include (1) the development of the next generation of the self-consistent and environment-dependent Hamiltonian (referred as SCED-LCAO) beyond group IV elements and the prediction of low dimensional boron structures based on icosahedra B12 using SCED-LCAO; (2) Li intercalation induced structural transition from black (A17 symmetry) to blue (A7 symmetry) phosphorene with and without pressure; (3) high performance Li-ion batteries with phosphorene as anode materials for its ultra-fast Li diffusion and high Li capacity; (4) prediction of novel 2D materials of sandwiched phosphide binary compounds sheets with tunable bandgaps and anisotropic physical properties; (5) anisotropic and nonlinear mechanical properties in 2D materials; (6) blue and red shift of Raman spectra of layered black phosphorene under pressure, strain, and Li intercalation, (7) Li mobility in layered oxides, (8) superionic conductivity of sulfide-based conductors as the most promising solid electrolytes for next-generation of all-solid state batteries, (9) the adsorption mechanism of 2D WS2 as sensors; (10) the interface effects on planar heterostructures; and (11) electrostatic interlayer bonding effects on the 2D ionic-like heterostructures.
Video Proceedings of Advanced Materials

Upcoming Congress
